SCENIC Suite:
Unveil regulatory information
from single-cell data
SCENIC Suite is a set of tools to study and decipher gene regulation. Its core is based on SCENIC (Single-Cell rEgulatory Network Inference and Clustering) which enables you to infer transcription factors, gene regulatory networks and cell types from single-cell RNA-seq data (using SCENIC) or the combination of single-cell RNA-seq and single-cell ATAC-seq data (using SCENIC+).
Read the original SCENIC paper (Aibar et al., 2017) Read the SCENIC protocols paper (Van de Sande et al., 2020) Read the SCENIC+ paper (Bravo and De Winter et al., 2023)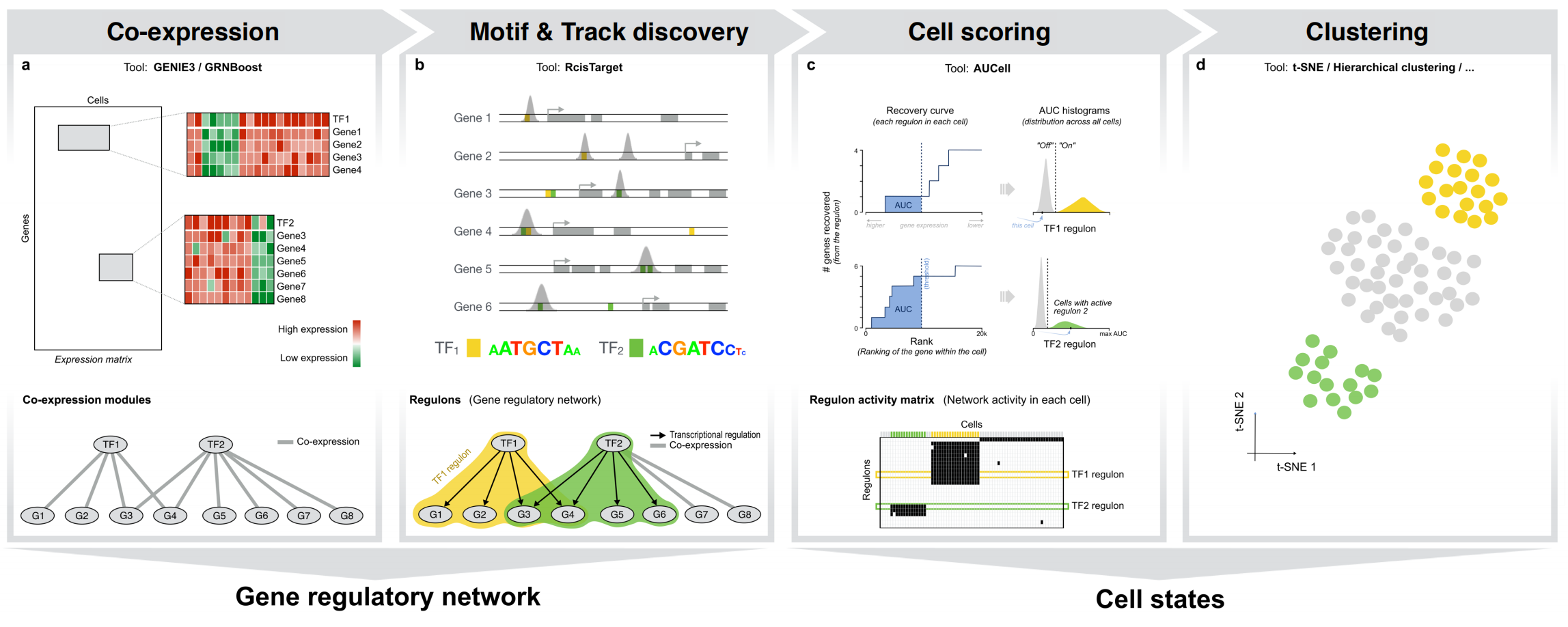
How to run ?
SCENIC for single-cell RNA-seq data:
There are currently implementations of SCENIC in R and in Python. If you don’t have a strong preference for a specific version, we would recommend starting with the SCENIC protocol repository, which contains Python/Jupyter notebooks to easily run SCENIC interactively For a more automated solution we recommend the VSN Pipelines (Nextflow DSL2; highly recommended for running it in batch or big datasets).
The output from any of the implementations can then be explored either in R, Python or SCope (a web interface).
SCENIC+ is implemented as a python package available at the SCENIC+ repository tutorials and documentation is available on the SCENIC+ readthedocs page.
SCENICprotocol
Python/Jupyter notebooks illustrating how to run pySCENIC alongside a basic ‘best practices’ expression analysis for single-cell data. Includes a basic implementation in Nextflow DSL1.
VSN-Pipelines
A Nextflow DSL2 workflow, which provides a method for running pySCENIC (and 100x SCENIC) seaminglessly on a high-performance cluster.
SCENIC+
SCENIC+ is a python package for enhancer-driven Gene Regulatory Network inference from combined single-cell chromatin accessibility and gene expression data.
SCENIC+ for combined single cell chromatin accessibility and gene expression
SCENIC+ is a tool which takes combined single-cell crhomatin accessibility and gene expression data as input to infere enhancer-drive gene regulatory networks. These are gene regulatory networks with enhancer support where we not only predit target genes of TFs but also target cis-regulatory regions. The package also contains modules to predict the effect of TF perturbations and to infer the differentiation effect of a TF (GRN velocity).
StarVisualize results in SCope
SCope is a visualization tool for single-cell datasets. The platform helps you to visualize and explore SCENIC results in an interactive way. It leverages an extended version of Loom developed by us, called LoomX. LoomX provides a defined metadata structure which allows multiple analyses to be stored within a loom file as well as a variety of other data types (such as cluster markers and regulons).
StarSupported species
We currently provide SCENIC databases for 3 species: Fly, Mouse and Human. For other species, check how to ‘Create your custom database’.


Create your custom database
SCENIC is a flexible method that allows you to create a database for custom species and/or with custom motif collections.
Motif enrichment, motif collections and motif2TF
The core of SCENIC is to use motif enrichment to link candidate TF regulators with candidate target genes. We use a large motif collection of position weight matrices that we collected from a wide variety of databases, and we are very grateful for the curators and developers of these databases. Here is an overview of these sources, alongside the credits and references to each motif database. We use motifs from different species, as DNA binding domains and PWMs are often conserved (this is similar to cis-bp). Motifs can be either directly annotated with a TF, or we link a motif to a TF because it is annotated by an orthologous TF in another species (e.g., a PWM from Drosophila dorsal is linked to human and mouse NFkB). A third layer of TF-motif links is provided by motif-motif similarities. For example, unannotated motifs in our collection can be linked to a TF by motif similarity to an annotated motif. This third layer is usually turned off by default.


Working with ChIP-seq tracks
SCENIC’s main approach is to search the putative regulatory regions of target genes for TF motif enrichment. This approach can be supplemented by the use of ChIP-seq track databases, which are based on direct experimental evidence of TF binding. Both direct and indirect binding are taken into account when generating pruned regulons. Track databases are currently available for Human and Drosophila.
Scalability
The first step of the SCENIC workflow is to infer potential transcription factor targets based on the gene expression data. This step is a computationally expensive task, exacerbated by increasing data sizes due to advances in high-throughput gene profiling technology. The Arboreto software library (Arboreto paper)) addresses this issue by providing a computational strategy that allows executing the class of GRN inference algorithms exemplified by GENIE3 on hardware ranging from a single computer to a multi-node compute cluster.
StarRobustness
SCENIC is a non-determinstic method due to the stochasticity that is inherent to the algorithms used to infer co-expression networks (GENIE3/GRNboost2). While the overall variability between runs is low, one way of increasing the confidence in the results is to run SCENIC multiple times and aggregate the generated regulons and TF-to-gene links. The computationally intensive work is facilitated by a Nextflow workflow (VSN-Pipelines) that can easily be submitted to any high-performance cluster.